Table of Contents
- GPU First, Then Balance the System
- What Balances an MD System?
- Why Do MD Engines Still Need CPU?
- How Much RAM Do MD Workflows Need?
- Is NVMe Storage Required for Production MD?
- Is Water Cooling Required for 24/7 MD?
- Your Workstation vs Cloud vs Cluster
- Common MD Workstation Mistakes
- Recommended BIZON MD Workstations
- Final MD Build Recommendations
Best CPU, GPU, RAM for Molecular Dynamics in 2026 [ Updated ]
Last verified: May 2026. System specs, GPU performance figures, and molecular dynamics software versions confirmed against official NVIDIA sources, GROMACS 2026.1 release notes, and bizon-tech.com.
For lab-scale MD simulations in 2026, the RTX 5090 is a leading consumer GPU option for AMBER, GROMACS, and OpenMM molecular dynamics workstations. A four-card water-cooled system runs four independent trajectories at sustained clock speed, with 128 to 256 GB ECC RAM and NVMe SSD scratch storage carrying most production work. Past 10 million atoms or for viral capsid NAMD, step up to H200-class VRAM, while everyone else wins by balancing the whole system. CPU, RAM, NVMe SSDs, and cooling each gate ns/day once the GPU stops being the bottleneck.

The 4-GPU RTX 5090 water-cooled MD workstation, hero configuration for the recommendations that follow.
ns/day measures simulated biological time processed in 24 hours. A 100 ns/day system finishes a 1 microsecond run in 10 days, and that throughput figure is the currency of every modern MD lab. Per the NAMD 3.0 release announcement and AMBER 24 docs, molecular dynamics has shifted into what NVIDIA calls the GPU-Resident era, where the simulation stays on the GPU instead of shuttling data back to the CPU every step. NAMD 3.0's GPU-Resident Mode reports a multi-fold gain over GPU-offload on the same hardware.
NVIDIA Developer on the High Performance Computing (HPC) software stack that powers modern MD engines, useful context for the GPU-Resident era shift covered in this guide. NVIDIA Developer YouTube channel.
The GPU-Resident shift compressed weeks of cluster queueing into desktop runtime, though picking the right GPU still leaves RAM, storage, cooling, and PSU sizing to gate sustained ns/day. Each spoke has a spec floor, and falling below it costs ns/day regardless of what the GPU can do. Balance decides the rest. Choose the GPU first, then balance every other spoke to match its bandwidth.
What Balances an MD System?
Five things gate ns/day in an MD workstation: GPU, CPU, RAM, NVMe SSDs, and sustained thermals. All five govern throughput because MD runs last days or weeks without a natural reset point. MD simulations differ from training-style workloads here, because there are no burstable windows. Every piece of the system has to sustain its throughput across the entire run. Sustained throughput governs everything.
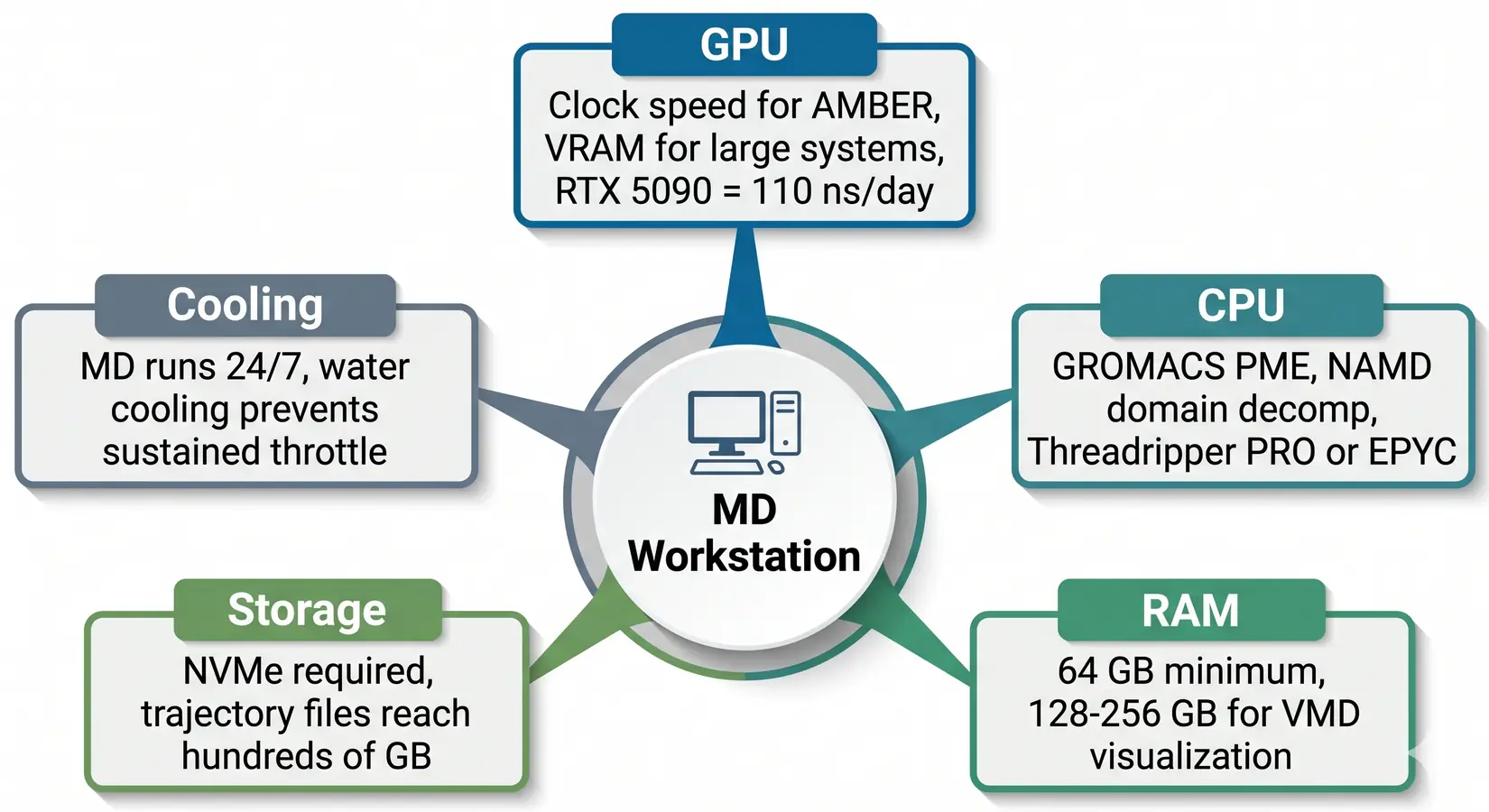
The balanced MD workstation. GPU dominates, but CPU, RAM, NVMe SSDs, and cooling all gate sustained throughput.
Drop any spoke a tier and the rig pays in lost ns/day. MD engines hand work between GPU, CPU, RAM, and NVMe SSDs every timestep. On our BIZON build floor, most labs under-spec the CPU when budget pours into more GPUs. GROMACS places PME and bonded-force work on CPU, so adding GPUs without matching CPU tier stalls at PME.
Why Do MD Engines Still Need CPU?
GROMACS, NAMD, and LAMMPS still run real work on CPU after the GPU-Resident shift, with bonded interactions, PME electrostatics, domain decomposition, and trajectory I/O all sitting on cores. Under-powered CPU bottlenecks the GPU, and core-count targets shift with the engine because each workload hits the CPU differently. The right CPU class depends on which engine the lab runs most of the week. The engine table below gives the per-engine working range.
- AMBER 24 - GPU-Resident, so CPU load is low. Clock speed matters more than core count.
- NAMD 3.0 - GPU-Resident since version 3, though communication and setup still touch the CPU. 8 to 16 cores per GPU is a typical working target.
- GROMACS 2026.1 - Per the GROMACS user guide on hybrid CPU-GPU execution, CPU runs PME and bonded forces in the default hybrid scheme. Most GROMACS practitioners want at least 16 CPU cores per GPU, with 24 to 32 for PME headroom.
- LAMMPS - Hybrid CPU and GPU by design. 16 to 32 CPU cores per GPU is the typical working range.
- OpenMM - GPU-oriented. A small CPU allocation of 4 to 8 dedicated cores is sufficient.
| Engine | CPU sensitivity | GPU scaling | VRAM sensitivity | Storage intensity |
|---|---|---|---|---|
| AMBER 24 | Low | Weak (one fast card wins) | Low to moderate | Moderate |
| NAMD 3.0 | Moderate | Strong | High at 10M+ atoms | High |
| GROMACS 2026.1 | High (PME on CPU) | Moderate | Moderate | Moderate to high |
| LAMMPS | Moderate to high | Strong | Moderate | Moderate |
| OpenMM | Low | Weak (one fast card wins) | Moderate | Moderate |
Methodology note. Bands reflect engine architecture and typical 100K to 1M atom production runs.
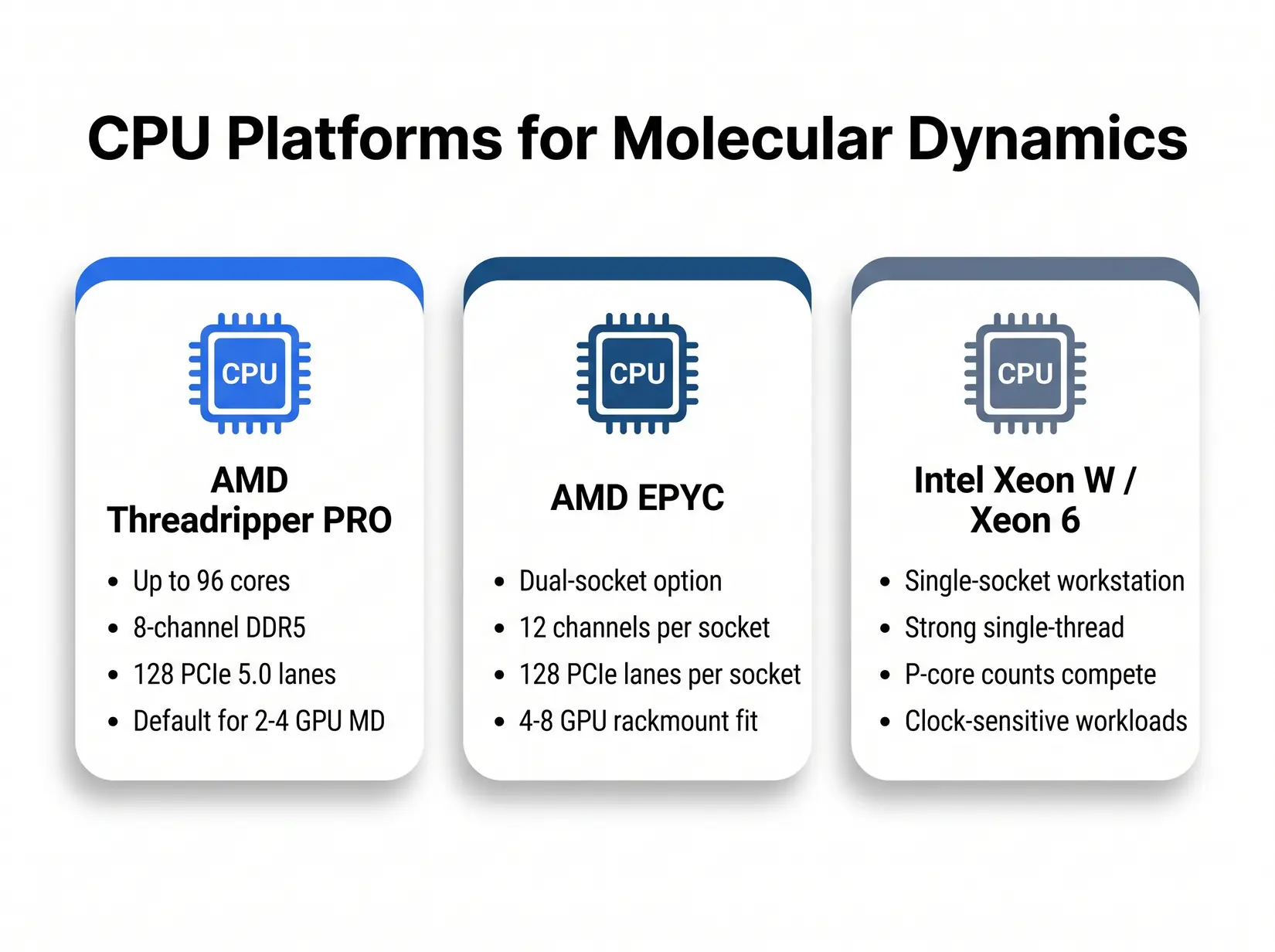
Three CPU platforms cover the MD workstation tier from desk-side to rackmount. Pick by workload pattern, not by brand.
All three platforms run every MD engine in this guide. GROMACS-heavy labs lean Threadripper PRO for its high core count and 8-channel memory bandwidth. NAMD single-socket work favors Intel Xeon W or Xeon 6 for clock-bound serial steps. EPYC takes dual-socket rackmount above 4 GPUs, where 12-channel-per-socket bandwidth and 256 PCIe 5.0 lanes give dense NAMD and GROMACS deployments room to scale, and under-provisioning CPU stalls GROMACS PME and leaves the GPU idle.
- AMD Threadripper PRO - Up to 96 cores on the 7000 WX series, 8-channel DDR5, and 128 PCIe 5.0 lanes. Default for 2 to 4 GPU MD workstations. This is the CPU in the BIZON ZX5500.
- AMD EPYC - Dual-socket configurations unlock 12-channel memory per socket and 128 PCIe 5.0 lanes per socket per the EPYC 9004 and 9005 specs. Pick EPYC for dense NAMD and GROMACS above 4 GPUs.
- Intel Xeon W and Xeon 6 - Solid single-socket workstation option. Strong single-thread performance for serial NAMD and LAMMPS, and Xeon 6 P-core counts hold their own against Threadripper PRO on clock-sensitive workloads.
Rule of Thumb
A common starting point: roughly 16 to 32 modern CPU cores per GPU for CPU-heavy MD engines (GROMACS 2026.1, LAMMPS), 8 to 16 allocated per card for NAMD, and 4 to 8 dedicated cores for GPU-Resident engines (AMBER, OpenMM). Tune from there based on PME balance, system size, and engine version. A four-GPU GROMACS rig typically wants at least a 64-core Threadripper PRO or dual EPYC. An AMBER-only rig needs far fewer.
MD simulations are more CPU-sensitive than broader scientific computing and data science workloads, since GROMACS hands the GPU a batch every timestep while the CPU is still computing PME and bonded forces, creating measurable ns/day penalties when under-sized. MD engines keep the CPU active at every timestep, unlike LLM inference where the CPU idles through long compute passes. CPU sizing sets the floor on how many trajectories the system can feed, and RAM closes the ceiling. CPU is the gatekeeper.
How Much RAM Do MD Workflows Need?
64 GB is the floor for dedicated MD work, and 128 to 256 GB covers most production trajectories. A lab running four GROMACS trajectories holds four sets of topology, coordinate, and force arrays in system RAM, plus the analysis pipeline pulling each trajectory back off disk. ECC catches the bit flips that silently corrupt a week-long trajectory. Non-ECC keeps going as if nothing happened.
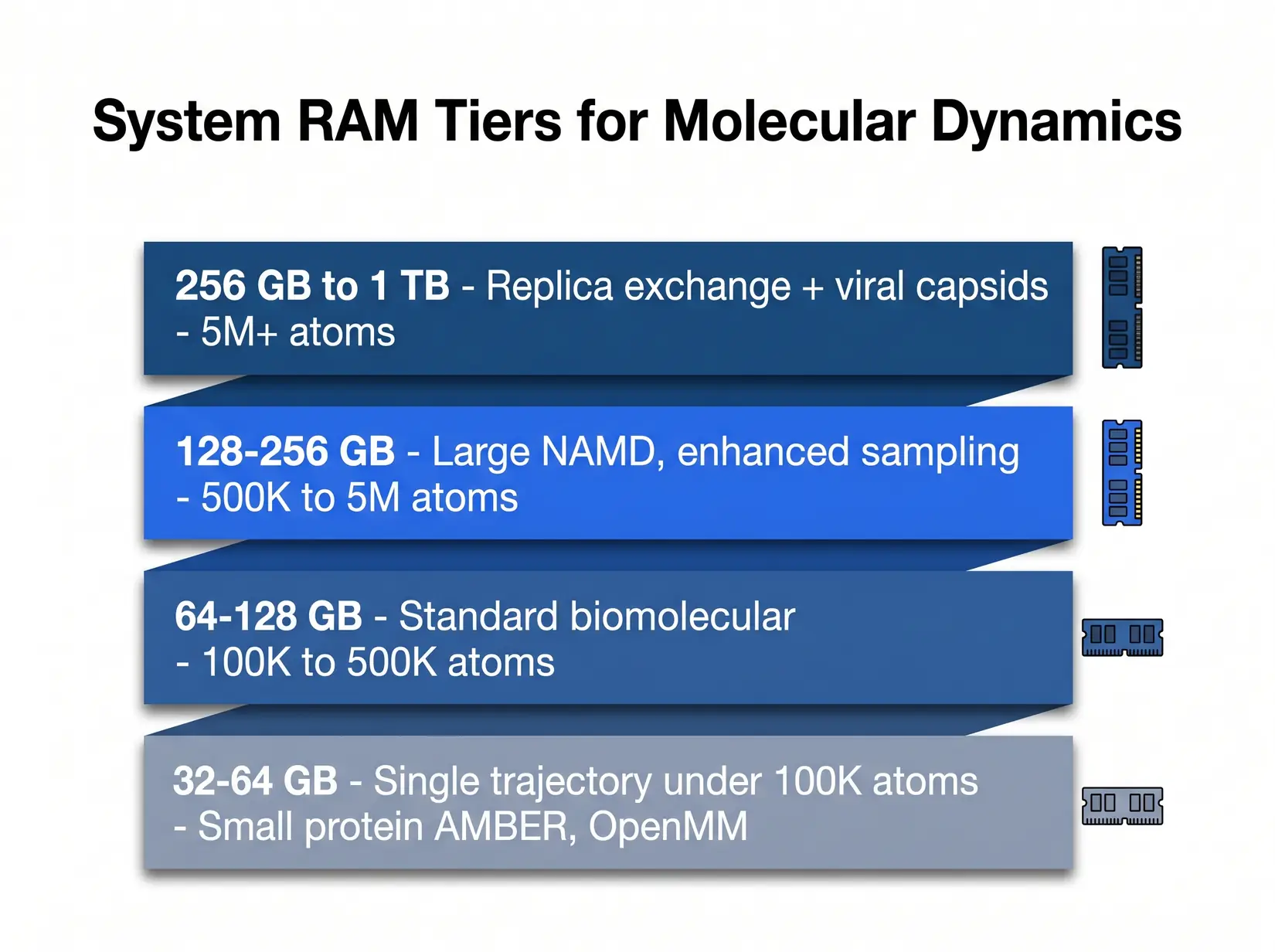
RAM scales with system size and replica count. Most production MD labs land in the 128 to 256 GB tier.
The ensemble column is where replica exchange and parallel tempering expose the hidden multiplier. A 128 GB rig that handles a single 500K atom trajectory cleanly chokes on an 8-replica exchange of the same system. Each replica holds its own in-memory state, so memory cost multiplies by that count. Size against the single-trajectory column first, then check the ensemble column.
| System Size | Single Trajectory RAM | Four-trajectory Ensemble or Replica Exchange RAM | Typical Workload |
|---|---|---|---|
| Under 100K atoms | 32 GB | 64 to 128 GB | Small protein AMBER, OpenMM |
| 100K to 500K atoms | 64 GB | 128 to 256 GB | Standard biomolecular AMBER, GROMACS, NAMD |
| 500K to 5M atoms | 128 GB | 256 to 512 GB | Large NAMD, enhanced sampling |
| Over 5M atoms | 256 GB | 512 GB to 1 TB | Viral capsid, whole-cell organelle |
Methodology note. These ranges reflect working memory for trajectory files, analysis, and concurrent replicas. VMD loads full trajectories into RAM during analysis. Size for peak pipeline load, not steady-state simulation.
Memory channel population matters as much as total capacity, since filling all eight slots on a Threadripper PRO or all 12 per socket on an EPYC gives the CPU the bandwidth to feed multiple GPUs simultaneously. Half-populating starves the CPU and shows up as lower ns/day even when the GPU headroom looks fine. Populate every channel on multi-GPU rigs, even when capacity looks sufficient. Channel count is throughput. Spare channels waste it.
Is NVMe Storage Required for Production MD?
NVMe SSDs are strongly recommended for four-trajectory parallel runs because SATA bottlenecks under checkpoint and analysis bursts. A GROMACS or AMBER run can generate hundreds of GB of trajectory data. PCIe Gen 5 drives like the Samsung 9100 Pro (14,800 MB/s) and WD_BLACK SN8100 (14,900 MB/s) post sequential reads 25 times the 550 MB/s SATA ceiling. Gen 5 is the floor.
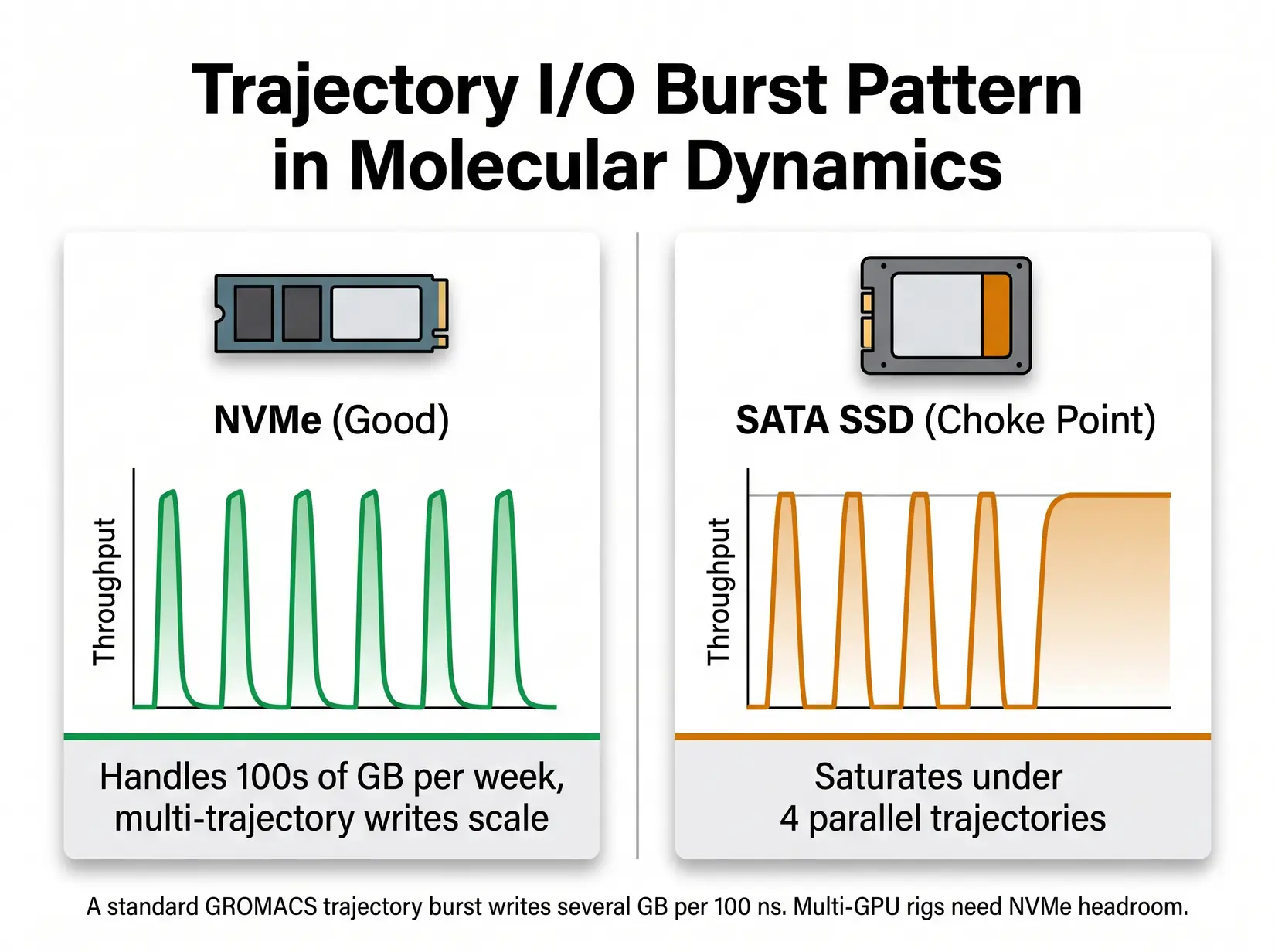
MD trajectory writes are bursty. NVMe SSDs absorb the bursts. SATA SSDs commonly become a bottleneck under multi-trajectory checkpoint and analysis workloads.
Three write patterns decide the NVMe storage floor. Sustained trajectory output runs continuously, while periodic full-state checkpoints hammer IOPS in concentrated bursts. The analysis phase pulls full trajectories back into memory for VMD or MDAnalysis. Match drive class to the heaviest pattern the lab runs, since that pattern sets the floor.
- Production trajectory output - A 500K-atom GROMACS run can generate several GB of .xtc data per 100 ns, and multi-GPU rigs accumulate active trajectory data quickly.
- Checkpoint and restart - NAMD and AMBER write full state checkpoints every N steps. On large NAMD runs those files reach multiple GB and are rewritten repeatedly, so IOPS matter as much as throughput.
- Analysis phase - VMD and MDAnalysis load full trajectories into memory. Slow reads cost analyst hours, and those hours compound across a multi-trajectory rig.
Costly Mistake
Buying a multi-GPU MD workstation with a single SATA SSD as the only fast disk. Four parallel trajectories on 4 GPUs will saturate SATA bandwidth during checkpoint and analysis. The disk budget should track the GPU count, not the single-workstation legacy pattern.
For single-GPU workstations, a 2 to 4 TB NVMe drive is the starting point and handles trajectory output and checkpoints without backpressure. For 2 to 4 GPU rigs running ensembles, pair a fast NVMe tier with a slower archive tier. For 4 to 8 GPU department-scale systems, 4 to 8 TB of fast SSD plus 20 TB or more of bulk archive covers most pipelines. We see eight-trajectory rigs fill 4 TB before the week ends, so size to campaign length, not single-trajectory throughput.
Is Water Cooling Required for 24/7 MD?
Water-cooled BIZON multi-GPU chassis hold sustained GPU clocks through multi-day MD production runs. Air-cooled four-plus GPU consumer builds may throttle under the same conditions. Sustained clocks separate the two. Per NVIDIA's RTX 5090 datasheet (575W) and the RTX PRO 6000 at 600W, thermal density inside a 4-GPU chassis climbs fast, and cooling architecture decides whether the GPUs hold clock or throttle.
BIZON on liquid-cooled multi-GPU chassis running four cards under sustained MD production load. BIZON Technology YouTube channel.
PSU sizing is the other failure mode that gets missed on multi-GPU MD. On a four-RTX-5090 workstation, GPU TDP alone reaches 2,300W and system draw runs higher. Multi-rail 80 Plus Gold or better PSUs, sized to the full chassis configuration, absorb the transient spikes when every GPU hits a kernel boundary simultaneously. Single-rail builds trip mid-run at that envelope. Sizing matters here.
From the BIZON Build Floor
On our build floor, a four-GPU RTX 5090 air-cooled chassis runs with fans pinned near max under sustained MD load, and the inner cards may throttle under continuous 24/7 production. The ZX water-cooled equivalent holds sustained GPU clocks across week-long runs and runs noticeably quieter. The compound cost of throttle is not a single event, it is the same pattern repeating through every multi-day campaign.
Water-cooled costs more upfront and pays back in sustained ns/day on 24/7 production. Air cooling fits labs that run trajectories during business hours and rest the system overnight. Labs targeting seven-day unattended runs need water cooling and local storage from the start. The cooling and cloud-versus-own decisions are linked at that duration. Plan both together.
Your Workstation vs Cloud vs Cluster
A workstation pays full price up front and typically runs three to five years across daily MD production. The hardware is yours to configure and optimize against your pipeline. Queue waits disappear. Data stays in-lab.
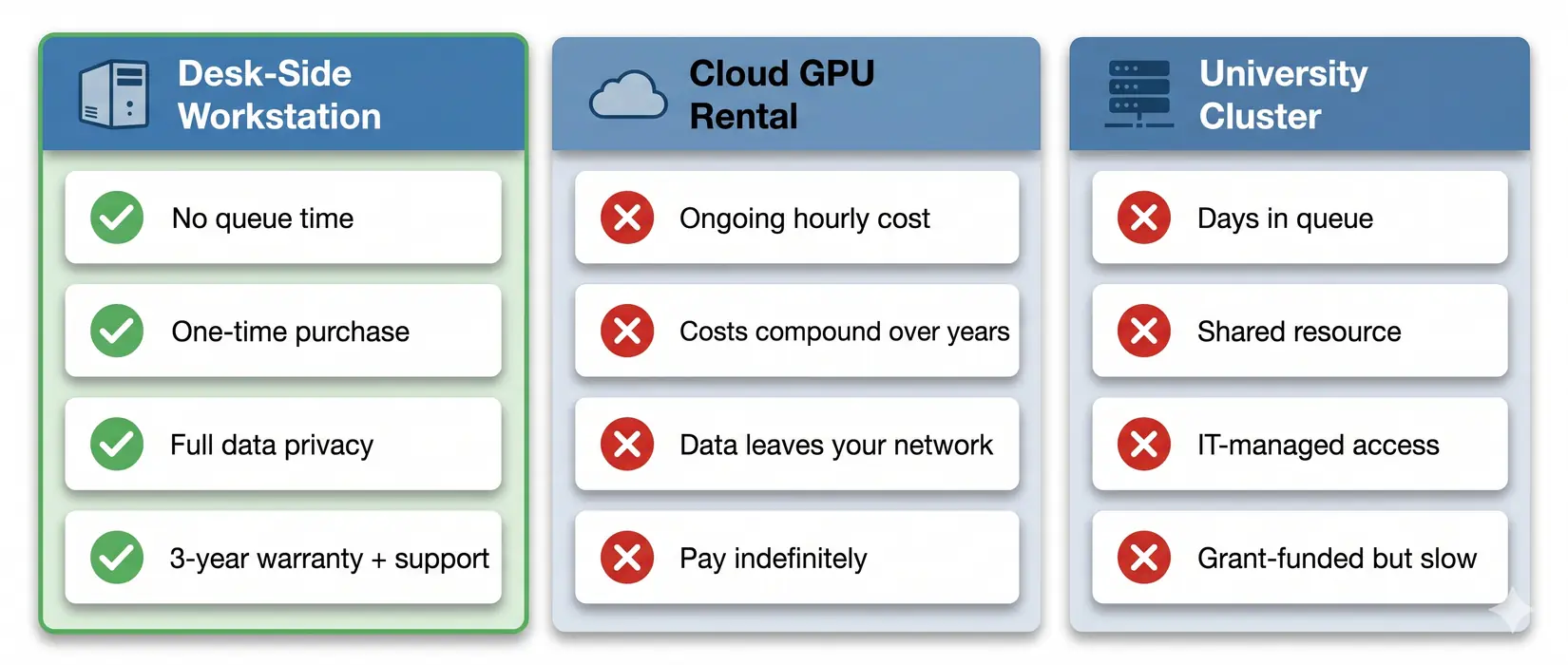
Queue time, data privacy, and long-run cost all favor a local workstation for sustained MD workloads.
A cluster trades capital cost for queue time and shared-tenancy constraints. Cloud rental meters every trajectory, checkpoint, and analysis pass. Owned hardware stops the meter and returns full scheduling control. For labs with daily production, ownership wins over a 3 to 5 year cycle.
On our build floor, most MD workstation regrets trace to the same five mistakes. The common thread is a lab that let CPU, RAM, or cooling drift a tier down while the GPU took the budget. Data stays in-lab. These mistakes surface on pre-sales calls.
Common MD Workstation Mistakes
The biggest MD system mistake is under-sizing CPU, RAM, or storage relative to GPU count. On our build floor, fixing the chassis after a build typically runs 3 to 5 times the original tier-down savings. GPU-choice mistakes (H100 for AMBER, 24 GB for 50M atoms) live in the MD GPU buyer guide. All five below are system-level and fixable at spec time.

System-build mistakes we see on real customer calls.
Five mistakes come up consistently across pre-sales calls and post-build support tickets. Each is a tier-down decision on one component that bottlenecks the rest of the chassis. CPU, storage, and RAM together account for most post-build correction requests we handle. The fix costs more than the right tier would have from the start. Spec right the first time.
- Ignoring CPU for GROMACS - PME and bonded force calculations still run on CPU cores. A weak CPU bottlenecks the whole pipeline.
- No NVMe storage budget - GROMACS .xtc and AMBER NetCDF trajectories generate hundreds of GB. Spinning disks kill I/O. A single SATA SSD chokes under four parallel trajectories.
- Under-sized RAM for replica exchange - Replica exchange and parallel tempering multiply in-memory state by the number of replicas. A 64 GB system runs fine for a single trajectory but locks up on an 8-replica exchange across a 500K atom system.
- Air-cooled 4-GPU consumer chassis for 24/7 max-load production - Four RTX 5090s in an airflow chassis may throttle under sustained 24/7 MD load, and lost throughput compounds across a production week. Water cooling belongs in the chassis decision when four consumer GPUs run continuous max-load production.
- Single-PSU builds for multi-GPU Blackwell - Transient spikes on four RTX 5090s trip nameplate-matched single-PSU rigs mid-run. Multi-GPU MD systems need redundant 80 Plus Gold, Platinum, or Titanium PSUs sized to the full chassis configuration.
Budget the system, not just the GPU. On a 24/7 MD run, every spoke that drops a tier costs ns/day.
The tiers below pair each chassis with its matching profile. Spec the chassis to the GPU count first and the workload pattern second. Each tier maps to a specific atom-count range and engine combination. Start with the engine table and atom count from the sections above, then match to the tier.
Recommended BIZON MD Workstations
BIZON covers three MD tiers, from a single-researcher workstation up to an 8-GPU SXM5 server with NVLink fabric for whole-cell NAMD work. Match atom count and engine to the VRAM ceiling, then confirm CPU and RAM feed trajectories without becoming the bottleneck. For the GPU-by-engine breakdown, see the MD GPU buyer guide. Each tier ships with the MD stack pre-installed and validated against published benchmarks.
Individual Researchers (1 to 2 GPU)

BIZON X3000 G2 Desktop Workstation
- Best for: Single-researcher AMBER, OpenMM, or GROMACS up to about 1M atoms on AMD silicon
- GPUs: Two GPU slots, full RTX Blackwell and Ada workstation lineup (no datacenter SXM or HBM)
- VRAM: Up to 192 GB total (two RTX PRO 6000 at 96 GB ECC each)
- CPU: AMD Ryzen 9000 (9900X or 9950X, up to 16 cores)
- RAM: Up to 256 GB DDR5, dual-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 100 Gbps InfiniBand

BIZON V3000 G4 Desktop Workstation
- Best for: Same single-researcher MD profile as the X3000, on Intel silicon
- GPUs: Two GPU slots, full RTX Blackwell and Ada workstation lineup (no datacenter SXM or HBM)
- VRAM: Up to 192 GB total (two RTX PRO 6000 at 96 GB ECC each)
- CPU: Intel Core Ultra 9 (14, 20, or 24 cores)
- RAM: Up to 192 GB DDR5, dual-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 100 Gbps InfiniBand
Drug Discovery Labs (2 to 4 GPU)

BIZON X5500 G2 Workstation
- Best for: Mid-tier flexible workstation for labs that mix AMBER, NAMD, and GROMACS across 2 to 4 GPUs
- GPUs: Up to four RTX Blackwell or Ada workstation cards
- VRAM: Up to 384 GB total (four RTX PRO 6000 at 96 GB ECC each)
- CPU: AMD Threadripper PRO (16, 24, 32, 64, or 96 cores)
- RAM: Up to 1,024 GB DDR5 ECC, 8-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 200 Gbps InfiniBand HDR

BIZON G3000 Gen2 Workstation
- Best for: Reliability-first ECC desktop for unattended multi-day production runs on Intel
- GPUs: Up to four RTX Blackwell or Ada workstation cards
- VRAM: Up to 384 GB total (four RTX PRO 6000 at 96 GB ECC each)
- CPU: Intel Xeon W-3500 (up to 60 cores) or Xeon W-2500 (up to 22 cores)
- RAM: Up to 1,024 GB DDR5 ECC Buffered, quad-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 100 Gbps InfiniBand EDR

BIZON R5000 Rackmount Workstation
- Best for: The X5500 workload in a 5U rackmount, for labs with a server room and a queue to replace
- GPUs: Up to four RTX Blackwell or Ada workstation cards
- VRAM: Up to 384 GB total (four RTX PRO 6000 at 96 GB ECC each)
- CPU: AMD Threadripper PRO (16, 24, 32, 64, or 96 cores)
- RAM: Up to 1,024 GB DDR5 ECC, 8-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 100 Gbps InfiniBand EDR

BIZON X8000 G3 Rackmount Server
- Best for: Dual-EPYC 2U rack for Hopper PCIe ECC at 4-GPU scale without water cooling
- GPUs: Up to four PCIe GPUs across RTX PRO Blackwell, RTX Ada, and Hopper PCIe (L40S, A100, H100 NVL, H200 NVL)
- VRAM: Up to 564 GB total (four H200 141 GB NVL)
- CPU: Dual AMD EPYC 9004/9005 (up to 192 cores per CPU)
- RAM: Up to 1,536 GB DDR5 ECC Buffered, 12-channel per CPU
- Connectivity: 10 GbE dual port (two RJ45), up to 200 Gbps HDR or 400 Gbps NDR InfiniBand
Department Scale (4 to 8 GPU)

BIZON ZX5500 Water-Cooled Workstation
- Best for: Water-cooled desktop tower for CPU-heavy GROMACS and LAMMPS pipelines at department scale
- GPUs: Up to seven water-cooled GPUs across RTX 5080/5090, RTX PRO 6000 Blackwell, and Hopper PCIe (A100, H100, H200)
- VRAM: Up to 987 GB total (seven H200 141 GB)
- CPU: AMD Threadripper PRO 7000 or 9000 (24, 32, 64, or 96 cores)
- RAM: Up to 1,024 GB DDR5 ECC, 8-channel
- Connectivity: 1 GbE built-in plus Wi-Fi/Bluetooth, up to 25 GbE or 100 Gbps InfiniBand EDR

BIZON ZX9000 Water-Cooled Server
- Best for: 8-GPU water-cooled server for replica exchange and viral-capsid NAMD where PCIe peer-to-peer scales adequately
- GPUs: Up to eight water-cooled GPUs across RTX PRO 6000 Blackwell and Hopper PCIe (A100, H100 NVL, H200 NVL)
- VRAM: Up to 1,128 GB total (eight H200 141 GB NVL)
- CPU: Dual AMD EPYC 9004 or 9005
- RAM: Up to 3,072 GB DDR5 ECC Buffered, 12-channel per CPU
- Connectivity: 10 GbE dual port (two RJ45), up to 400 Gbps InfiniBand NDR

BIZON X9000 G3 HGX Server
- Best for: True 8-way SXM5 NVLink fabric for NAMD GPU-Resident Mode above 10M atoms when PCIe peer-to-peer is no longer enough
- GPUs: Eight H100 80 GB SXM5 or eight HGX H200 141 GB SXM5 (fixed SXM-only chassis)
- VRAM: Up to 1,128 GB total (eight HGX H200 141 GB SXM5)
- CPU: Dual AMD EPYC 9004/9005 or dual Intel Xeon Scalable 4th/5th Gen
- RAM: Up to 3,072 GB (AMD) or 4,096 GB (Intel) DDR5 ECC
- Connectivity: 10 GbE dual port (two RJ45), up to 400 Gbps InfiniBand NDR

BIZON X9000 G4 HGX Server
- Best for: Whole-cell NAMD and >30M atom systems where Hopper VRAM is no longer enough
- GPUs: Eight NVIDIA B200 192 GB SXM5 (fixed SXM-only chassis, 8 TB/s memory bandwidth per card)
- VRAM: 1,536 GB total (eight B200 192 GB SXM5)
- CPU: Dual AMD EPYC 9004/9005 or dual Intel Xeon Scalable 4th/5th Gen
- RAM: Up to 3,072 GB (AMD) or 4,096 GB (Intel) DDR5 ECC
- Connectivity: 10 GbE dual port (two RJ45), up to 400 Gbps InfiniBand NDR (Mellanox ConnectX-7)
Final MD Build Recommendations
The shortest route to production MD simulations is a system sized to the workload. The engine table and atom-count floors above collapse the decision to three numbers. Pick the MD engines you run, the largest atom count you target, and the parallel trajectory volume you need. Those three drive every chassis decision below. Start with the engine.
From our experience at BIZON, labs that get the most ns/day per dollar match every spoke to GPU count. AMBER takes CPU clock speed, while GROMACS PME needs CPU cores. Water cooling holds sustained four-card production clocks. Clocks matter through the campaign. Every MD system ships with BizonOS preloaded.
Configure your MD build through BIZON's molecular dynamics workstation and server lineup, from one-GPU desk-side rigs to eight-GPU rackmount density. Each tier ships with NAMD, GROMACS, LAMMPS, and VMD validated as GPU-accelerated containers. Our team configures every build to the specific engine mix and atom-count targets the lab runs. Spec the system to the engine and atom count today, with headroom for the next class of simulation.
BIZON manufactures the workstations and servers discussed in this article. Benchmark figures and product specifications reflect published vendor sources and BIZON build-floor experience. Our editorial recommendations follow the engine-first, workload-first analysis shown above. They are not constrained by inventory or commercial considerations.