Table of Contents
- The MD Engine Picks the GPU First
- Quick GPU Picks by MD Software
- Which GPU Specs Matter for MD?
- RTX 5090 vs H100 vs H200 for AMBER MD
- The VRAM Cliff: 32, 80, 141 GB
- Which GPU Fits Your MD Engine?
- Consumer or Datacenter GPU for MD?
- Common GPU Mistakes for MD Workloads
- BIZON MD Workstations by GPU Tier
- Single-GPU Starting Points
- Drug Discovery and Mid-Size Labs
- Department Scale and NAMD HPC (4 to 8 GPU)
- Getting Started With Your MD GPU
The Best GPU for Molecular Dynamics: A Comprehensive Guide 2026 [ Updated ]
Last verified: 2026. Specs and benchmark figures confirmed against official sources.
In 2026, the RTX 5090 is a strong choice for many lab-scale molecular dynamics (MD) workloads, especially single-GPU AMBER, Python-scripted OpenMM, and ensemble GROMACS screening. That answer reverses the last two buyer-guide generations. AMBER's pmemd.cuda kernel (AMBER's CUDA-native GPU-resident simulation engine) is one of the most mature single-GPU MD kernels in the field, and it rewards clock speed over SM count. Blackwell's clock uplift over Hopper converts directly into ns/day, so a single consumer card commonly out-simulates a datacenter card that costs several times as much on AMBER STMV benchmarks and across GROMACS and OpenMM at lab-scale atom counts. The right answer depends less on datacenter-versus-consumer than on which MD engine runs most of the week.

The GPU picks below assume the system around them is sized correctly. A mis-specced CPU or a PCIe layout that starves the top GPU slot silently caps production ns/day regardless of the card. The sections that follow walk each engine's GPU-tier reasoning. For the matching CPU tier, memory-channel population, NVMe storage, cooling, and PSU sizing, see Best Hardware for Molecular Dynamics Simulations in 2026.
Quick GPU Picks by MD Software
For most academic MD labs in 2026, a Blackwell consumer card with 32 GB handles everyday pmemd.cuda, ensemble GROMACS, and Python-scripted OpenMM pipelines. Large-scale NAMD work or biomolecular systems above roughly 10 million atoms typically justify stepping up to Hopper-class GPUs with larger VRAM and interconnect advantages.
Engine choice decides GPU tier more reliably than any other lever in MD hardware. The table pairs each engine with its best-fit GPU, the VRAM tier, and the reason. Read the "Why" column first. That's the part most buyers miss starting from a spec sheet instead of their lab's actual engine.
| Engine | Sweet-spot GPU | VRAM tier | Why |
|---|---|---|---|
| AMBER 24 | RTX 5090 (single, or four for trajectories) | 32 GB | Clock-king kernel, no multi-GPU scaling per trajectory |
| NAMD 3.0 | RTX 5090 to four GPUs, H200 NVLink above 10M atoms | 32 to 141 GB | True multi-GPU scaling, GPU-Resident Mode |
| GROMACS 2026 | RTX 5090 (one per ensemble member) | 32 GB | Ensemble engine, CPU heavy, no cross-GPU scaling |
| OpenMM | RTX 5090 | 24 to 32 GB | Single-GPU Python pipelines, ML-potential ready |
| LAMMPS | RTX 5090 plus Threadripper PRO or EPYC | 24 to 32 GB | Hybrid CPU and GPU, CPU quality is the lever |
Pin down which engine your group actually runs most of the week, not which engine the original grant was written around. A lab that inherited an AMBER pipeline but now runs GROMACS for drug screening is a GROMACS shop. Both engines scale by running independent jobs across separate cards, not by NVLink within one job. Spending the budget on H100 SXM silicon for the old AMBER assumption pays for an interconnect neither engine ever uses, when the same money would have bought four RTX 5090s feeding four parallel trajectories. Start from the engine, not the card.
Engine-first, SKU-second is the only ordering that survives this decision.
Which GPU Specs Matter for MD?
Four GPU specs decide ns/day in MD: clock speed, VRAM ceiling, memory bandwidth, and multi-GPU interconnect. For conventional force-field MD, tensor cores and FP8 throughput are typically less relevant than those four specs.

Clock speed and VRAM determine fit for the most common MD workloads. Memory bandwidth and multi-GPU interconnect only earn their premium on the NAMD-at-scale exception. The bullets below name what each spec does inside the kernel.
- Clock speed - AMBER's pmemd.cuda kernel cares about clocks more than SM count. A Blackwell consumer card outpaces an H100 on STMV (Satellite Tobacco Mosaic Virus, the standard AMBER throughput benchmark) because the kernel cannot use a wider GPU to compensate for lower clocks.
- VRAM capacity - 32 GB covers the overwhelming majority of standard biomolecular simulations. Viral capsids, multi-protein assemblies, and whole-cell models above 10M atoms need 80 to 141 GB. The cliff is binary.
- Memory bandwidth - Per NVIDIA's datasheets, H200's 4,800 GB/s HBM3e against the RTX 5090's 1,792 GB/s GDDR7 matters once systems stress data movement instead of compute. Only NAMD GPU-Resident Mode sees the delta.
- Multi-GPU interconnect - PCIe peer-to-peer is fine at most lab scales. NVLink primarily earns its premium for NAMD above 10M atoms on more than 2 GPUs. Below that threshold, NVLink rarely earns its cost.
If you came from deep learning, the reflex is to compare FP8 tensor throughput. Suppress it. Force-field evaluation runs on standard CUDA cores in FP32 or FP64, and the FP8 tensor machinery that sells H100 LLM racks sits idle during pmemd or NAMD. Bring FP8 reasoning in only if the workstation pulls double duty as an inference box. See Best GPU for LLM Training and Inference in 2026. For shared ML pipelines, see Best GPU for Data Science in 2026.
RTX 5090 vs H100 vs H200 for AMBER MD
Per ProxPC's STMV benchmark, the H200 leads the tier at 135 ns/day and the RTX PRO 6000 follows at 121. The RTX 5090 at 110 ns/day versus H100 at roughly 90 is the inversion that reframes MD hardware shopping in 2026.
Read ns/day per VRAM GB first if production fits inside 32 GB, where the RTX 5090 leads. Read the legacy Quadro RTX 5000 column first if the workstation is still on Turing or Ampere, where every modern card clears at least a 3x uplift.
| GPU | ns/day | vs Legacy Quadro RTX 5000 | ns/day per VRAM GB | Best For |
|---|---|---|---|---|
| H200 141 GB | 135.03 | 6.75x | 0.96 | Massive systems above 10M atoms |
| RTX PRO 6000 96 GB | 121.56 | 6.1x | 1.27 | Large systems plus ECC reliability |
| RTX 5090 32 GB | 110.03 | 5.5x | 3.44 | Standard MD simulations, throughput-per-VRAM leader |
| H100 80 GB | ~90 | 4.5x | 1.13 | Multi-GPU NVLink scaling |
| RTX 6000 Ada 48 GB | 71.50 | 3.5x | 1.49 | Capable but being replaced |
Methodology note. STMV ns/day figures and the legacy Quadro RTX 5000 multiplier column come from ProxPC's published hardware comparison using the standard STMV production run in AMBER 24. The ns/day per VRAM GB column is derived by dividing each GPU's STMV ns/day by its rated VRAM capacity, not a ProxPC-published figure. H100 and H200 share identical compute silicon at roughly 989 FP16 TFLOPS per NVIDIA's datasheets, so the H200 win is bandwidth driven (4,800 GB/s HBM3e against H100's 3,350 GB/s).
The inversion surprises most first-time MD buyers expecting datacenter to beat consumer. AMBER's kernel is a masterpiece of single-GPU optimization, and Blackwell's clock uplift converts directly into ns/day because H100 trades peak boost for the 24/7 sustained envelope a datacenter card must hold. H100 earns its slot back once NAMD above 10M atoms enters the roadmap, where bandwidth and NVLink decide the run.
Key Takeaway
A four-GPU RTX 5090 workstation pushes roughly 440 ns/day of aggregate AMBER throughput across four independent trajectories, about 3.3x a single H200 on STMV. For drug discovery screening and replica exchange, this is the throughput-per-node sweet spot.
The VRAM Cliff: 32, 80, 141 GB
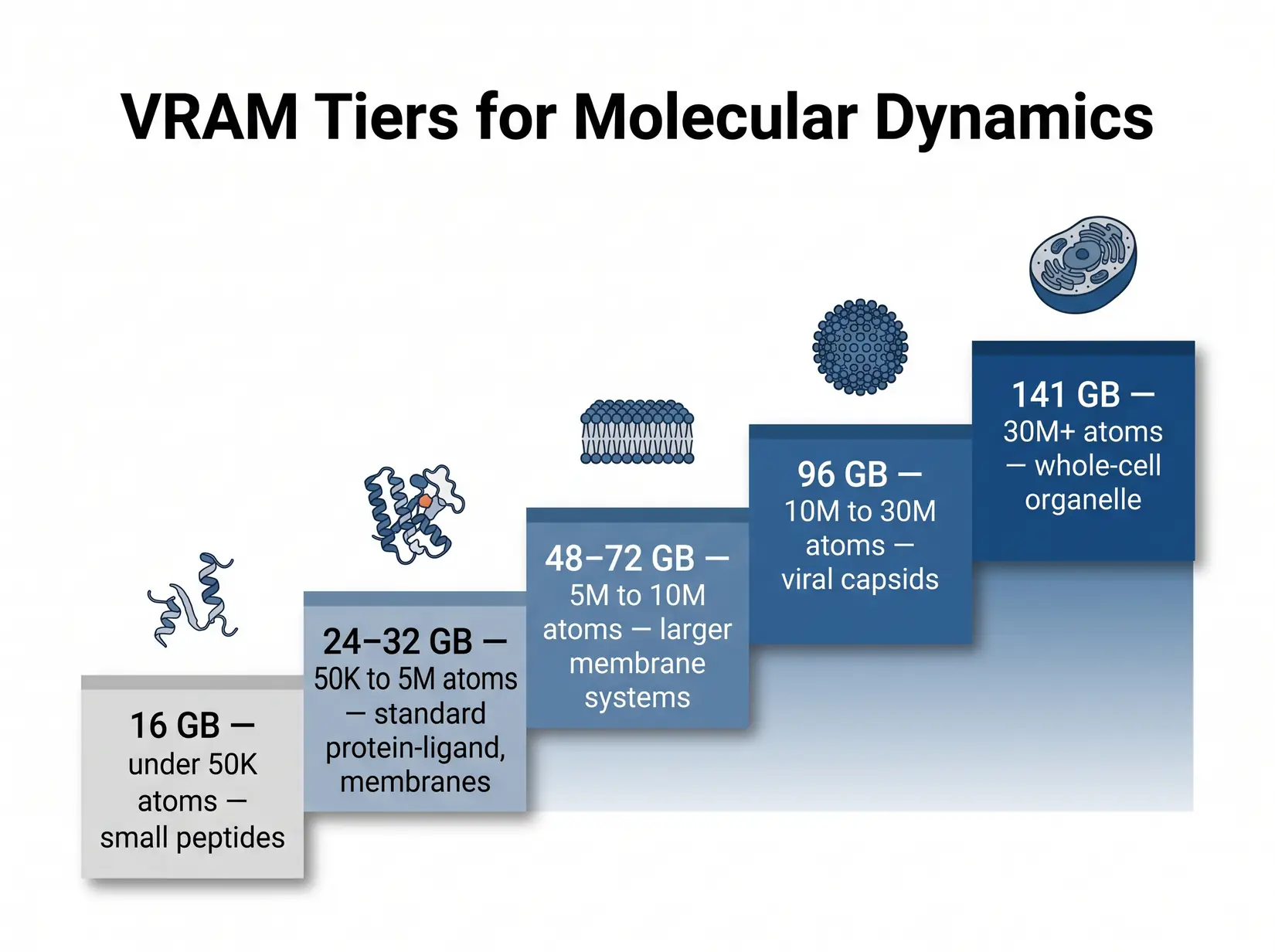
For AMBER pmemd.cuda, 32 GB handles standard biomolecular systems up to roughly 1 million atoms, STMV-class workloads. GROMACS offloads only non-bonded forces to the GPU and runs in a fraction of that VRAM, so the cliff is AMBER-specific at this tier. Viral capsids and whole-cell models cross into the 80 GB and 141 GB tiers, and the engine triggers an out-of-memory (OOM) crash at the boundary rather than slowing down. Size against the biggest system on the roadmap plus 20 percent headroom for restart files, replica buffers, and analysis scratch.
Non-ECC GDDR7 at 32 GB is fine for re-runnable trajectories. ECC HBM at 80 and 141 GB matters for 48-hour-plus unattended runs, where a bit flip silently corrupts the force-field state and surfaces at analysis when clustering fails. The failure pattern is particularly costly in MD because force-field state accumulates across millions of timesteps, so a bit flip at hour 12 of a 72-hour run goes undetected until the final clustering or RMSD analysis fails, writing off three days of compute.
| VRAM Tier | System size (atoms) | Examples | GPU options | ECC? |
|---|---|---|---|---|
| 16 GB | Small only (sub-megaatom) | Small peptides, ligand-only sims | RTX 5070 Ti, RTX 4080 (legacy) | No |
| 24 to 32 GB | Up to ~1M atoms | Most protein-ligand, membrane proteins, standard MD simulations | RTX 5090 | No |
| 48 to 72 GB | 5M to 10M | Larger membrane systems, multi-protein assemblies | RTX PRO 5000 (48 or 72 GB ECC) | Yes |
| 96 GB | 10M to 30M | Viral capsids, large complexes, ECC for unattended runs | RTX PRO 6000 (96 GB ECC) | Yes |
| 141 GB | 30M+ | Whole-cell organelle, massive NAMD systems | H200 | Yes |
Tier ranges trace ProxPC's published guidance for the AMBER engine, with HBM-tier cards becoming necessary at the viral-capsid threshold. NAMD GPU-Resident Mode runs heavier per atom because it keeps more of the neighbor-list structure in VRAM, so round NAMD system size up before mapping to a tier. Enhanced sampling methods (replica exchange, metadynamics, accelerated MD) multiply the baseline footprint by replica count plus collective-variable memory.
Costly Mistake
Buying 24 GB of VRAM for a 50 million atom system is a hard OOM, not a slowdown. The wrong VRAM tier turns the workstation into an expensive paperweight for that workload.
Which GPU Fits Your MD Engine?
Each MD engine rewards different GPU specs, and buying the wrong SKU for your software is the most common GPU mistake. The five major engines split into two patterns. AMBER, GROMACS, and OpenMM run fastest on a single clock-fast consumer card. NAMD at scale rewards bandwidth and interconnect, while LAMMPS rewards a strong CPU as much as the GPU.
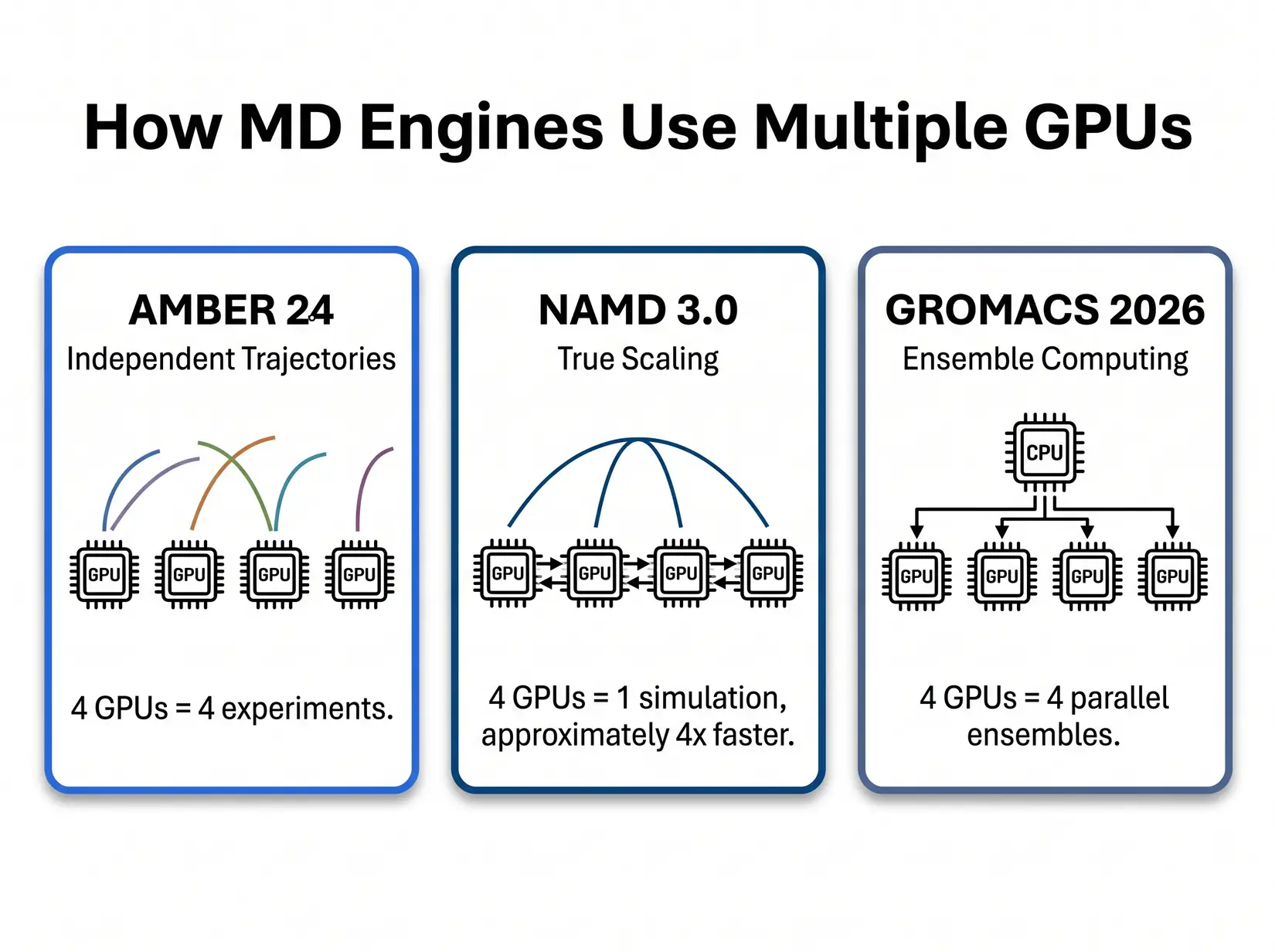
AMBER 24
AMBER 24's pmemd.cuda is purpose-built around a single-GPU design. The kernel, documented by Salomon-Ferrer, Case, and Walker in the pmemd.cuda papers (JCTC 2013), keeps the entire force-field pipeline on one device and avoids the CPU round-trip on every timestep. Adding GPUs to one AMBER run does nothing, and splitting a trajectory across devices falls back to the legacy multi-GPU path that loses the GPU-Resident speedup. Scale AMBER by running independent trajectories on separate GPUs.
Independent trajectories in parallel line up cleanly with replica exchange, enhanced sampling, and drug-discovery screening, and 32 GB handles standard biomolecular simulations. Labs crossing the 5M-atom line typically step to RTX PRO 6000 at 96 GB rather than pay for H100 silicon that pmemd.cuda never uses. Very few AMBER workloads need H200's 141 GB. The PRO 6000 also stays in PCIe topology, which is architecturally correct for AMBER. The pmemd.cuda single-GPU-first design means NVLink fabric adds zero AMBER throughput regardless of atom count, so there is no penalty for the cheaper interconnect.
NAMD 3.0
NAMD 3.0's GPU-Resident Mode is the only major MD engine where adding GPUs makes a single simulation faster. Per the NAMD 3.0 release notes, near-linear scaling is achievable on eight-A100 nodes, and NVLink on Hopper and datacenter Blackwell extends the curve onto bigger systems. That scaling only matters above roughly 10M atoms, below which force-exchange overhead eats the gain and a single fast GPU finishes the trajectory sooner than four split across the same kernel. Silicon budget earns its keep on whole-cell, viral-capsid, or ribosome-scale work, and gets wasted on 500K-atom protein simulations where a single RTX 5090 already saturates the engine.
The sweet spot for most academic NAMD labs is two to four RTX 5090s, stepping up to NVLink Hopper only after crossing 10M atoms. Viral capsids in that range need 80 to 141 GB on H100 or H200. Whole-cell models above 30M atoms push into H200-only territory for the 141 GB HBM3e neighbor lists.
NVLink Break-Even Decision Tree
NVLink earns its premium on exactly one MD workload. That workload is NAMD 3.0 GPU-Resident Mode, running on systems above 10M atoms, on more than two GPUs in a single node. Every other MD workload in 2026 runs on PCIe peer-to-peer just as well. Per NVIDIA's H100 datasheet, NVLink delivers roughly 900 GB/s aggregate bandwidth per Hopper GPU against PCIe 5.0 x16 at 128 GB/s bidirectional, and that premium is only worth paying when cross-GPU force exchange saturates PCIe.
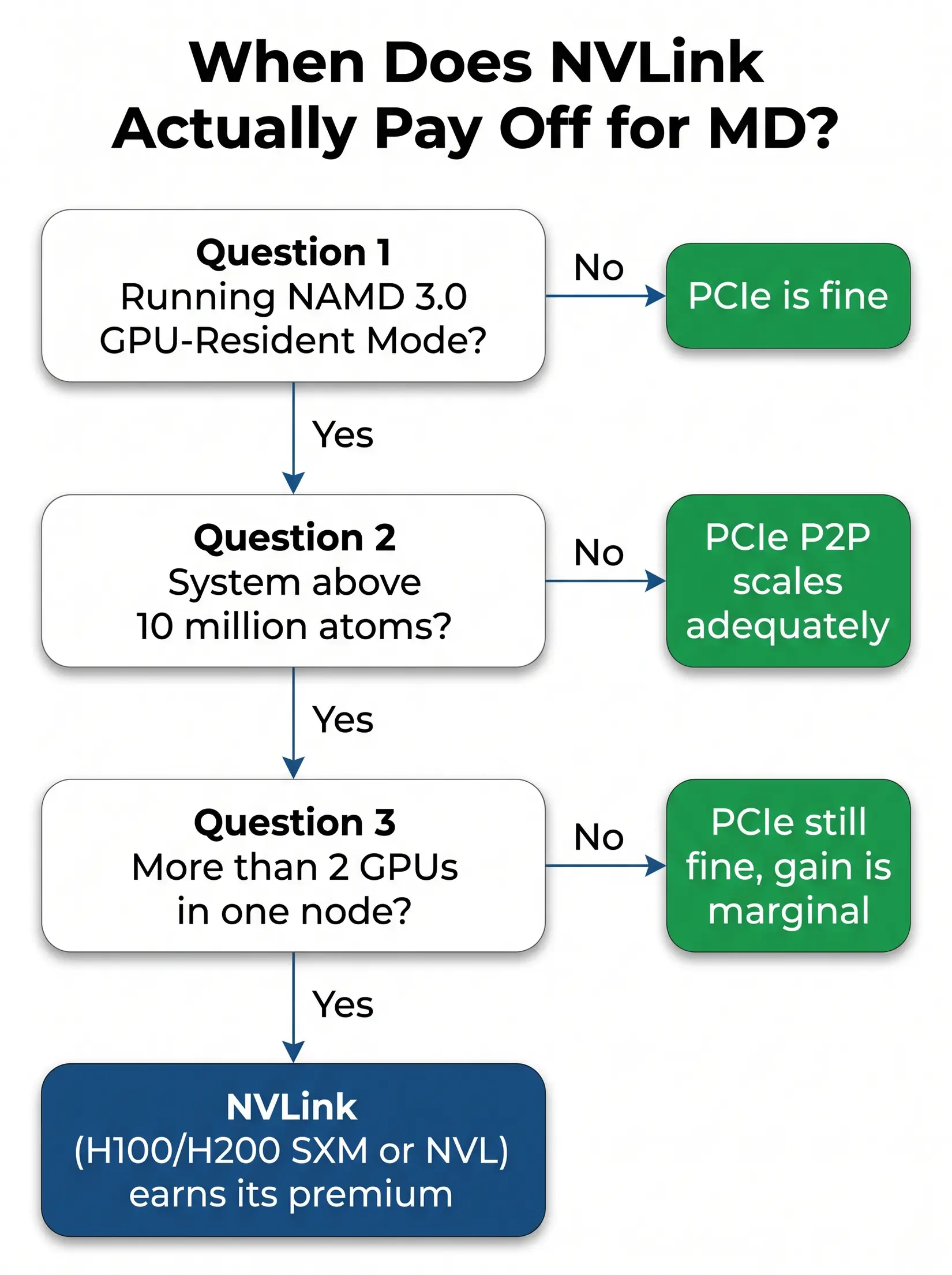
The flowchart above resolves into three dependent yes-or-no questions covering engine, atom count, and GPU count. Any "no" short-circuits the rest, and the answer becomes PCIe. The check takes under a minute against the lab's actual workload.
- Are you running NAMD 3.0 GPU-Resident Mode? If no, PCIe is fine. Stop.
- Is your system above 10 million atoms? If no, PCIe peer-to-peer scales adequately. Stop.
- Are you running on more than 2 GPUs in one node? If no, the NVLink bandwidth gain is marginal at that GPU count, and PCIe is still fine.
Yes to all three means NVLink earns its premium on H100 or H200 (SXM or NVL), because cross-GPU force exchange in the GPU-Resident kernel becomes the limit at that scale. The configurations that genuinely earn eight-way NVLink silicon are viral-capsid and whole-cell NAMD above 30M atoms. Everything else runs the same aggregate ns/day on four RTX 5090s at a fraction of the silicon cost.
GROMACS 2026, OpenMM, and LAMMPS
GROMACS 2026, OpenMM, and LAMMPS each top out at single-GPU sweet spots. RTX 5090 with a strong CPU companion is the right answer for all three, and engine choice dictates CPU tier rather than GPU tier. Spending the second-GPU budget on a stronger Threadripper PRO or EPYC instead delivers better throughput here.
GROMACS 2026 - GROMACS rewards ensemble computing, running multiple independent jobs rather than making a single job faster. It cares about CPU more than other GPU-Resident engines because of its Particle Mesh Ewald (PME) and bonded-force pipeline. 32 GB per GPU handles standard runs, and the sweet spot is four RTX 5090s with a Threadripper PRO or EPYC feeding them. Per the GROMACS 2026 release highlights (current as of April 2026, succeeding the 2026.0 January 19 GA), this cycle adds AMBER force-field ports, a full AMD HIP backend, and Neural Network Potentials.
Once the GROMACS kernel is tuned, GPU feeding is the bottleneck, so the lever a lab has is CPU clocks and PCIe topology rather than a second GPU.
OpenMM - OpenMM rewards GPU-oriented design. The GPU does nearly all the computation and the CPU coordinates, making it excellent for custom force fields, Python-scripted pipelines, and ML-potential workflows on TorchMD or openmm-torch. GPU acceleration commonly reaches 20x or more over a high-end desktop CPU on standard MD force fields, so single GPU is enough. 24 to 32 GB on an RTX 5090 covers it.
LAMMPS - LAMMPS rewards a hybrid CPU and GPU approach. GPU handles non-bonded interactions, CPU handles bonded forces, and CPU quality matters more here than in any other common MD engine. For polymer simulation, materials science, or non-biological MD, budget more system into the CPU. The sweet spot is an RTX 5090 paired with a strong Threadripper PRO or EPYC.
Consumer or Datacenter GPU for MD?
Several affordable cards beat one flagship SKU for most MD labs. Only NAMD above 10M atoms inverts it. AMBER and GROMACS are embarrassingly parallel at the system level, so four consumer cards deliver four simultaneous experiments at roughly the silicon-tier cost of a single datacenter card. NAMD is the exception because GPU-Resident Mode distributes one simulation across multiple GPUs, making interconnect bandwidth the only place that moves the ns/day needle on a single trajectory.
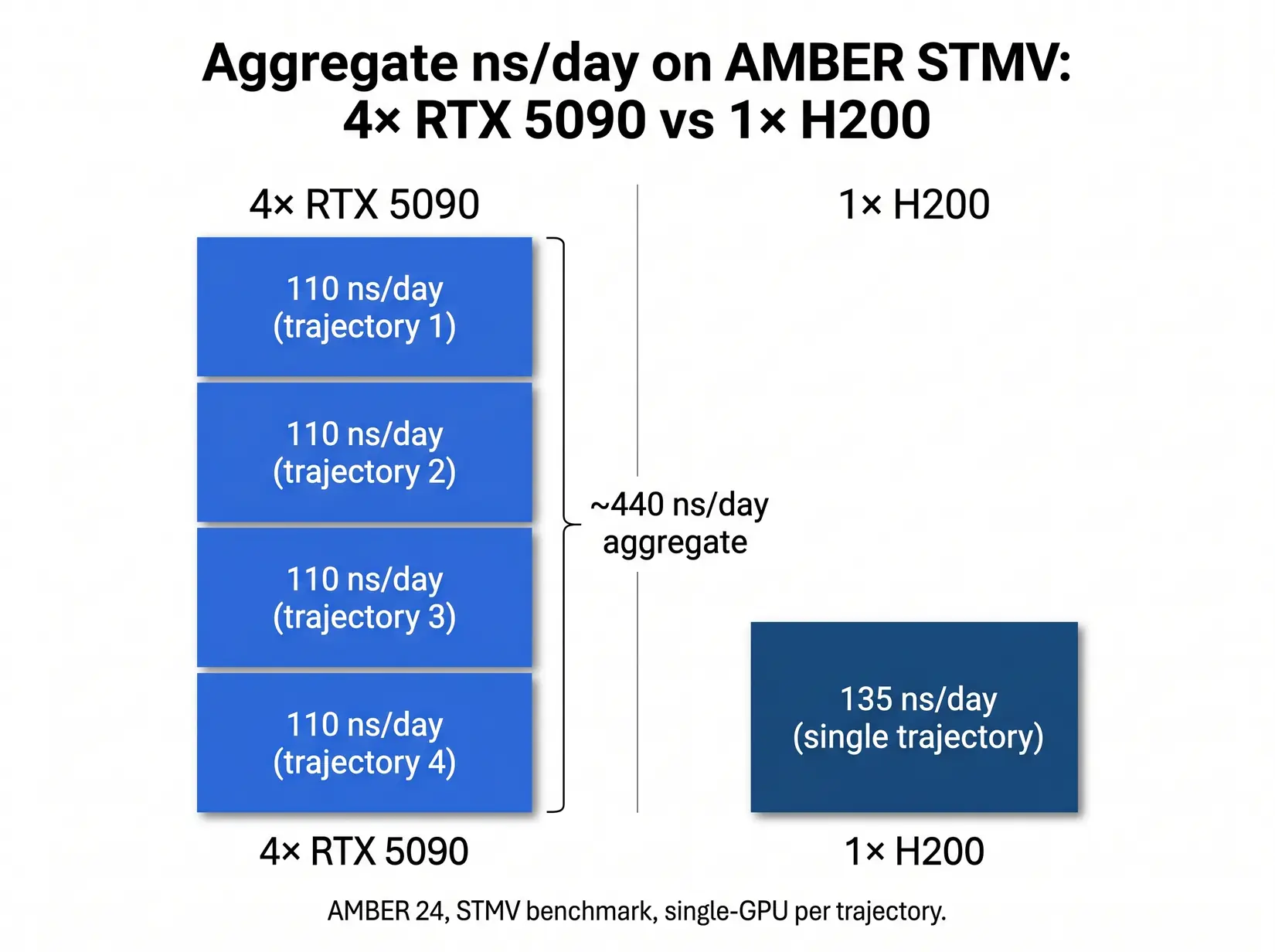
The bar chart makes the aggregate case. Four independent AMBER trajectories on consumer cards deliver 3.3x the total ns/day of a single H200 at a fraction of the datacenter silicon cost. The table maps each hardware pattern to the engine workload that fits it, from single-priority NAMD at the H200 row through parallel AMBER and GROMACS screening at the four-RTX-5090 row to the mixed-workload core facility option at roughly 540 ns/day across four independent H200 trajectories (AMBER does not use NVLink, so the 540 figure is four independent runs, not a single scaled simulation).
| Configuration | STMV ns/day (engine pattern) | Aggregate throughput pattern | Best fit |
|---|---|---|---|
| Single H200 (one trajectory) | 135 (AMBER), scales linearly (NAMD) | One fast simulation | NAMD above 10M atoms, single-priority workflow |
| Four RTX 5090 (four trajectories) | 4 x 110 = 440 aggregate (AMBER) | Four independent simulations | Drug discovery screening, replica exchange, GROMACS ensembles |
| Four H200 (independent trajectories, AMBER does not use NVLink) | ~540 (AMBER, 4 independent runs), scales near-linearly (NAMD) | Four independent simulations OR one NAMD trajectory at scale | Whole-cell NAMD, mixed-workload core facilities |
Methodology note. Aggregate figures derive from the per-GPU STMV numbers above, multiplied by GPU count for engines with no cross-GPU scaling, validated against NVIDIA's NAMD v3 scaling documentation. PCIe 5.0 lane distribution affects sustained multi-trajectory throughput, see the MD system configuration article.
Run the per-watt and per-VRAM-GB math and the picture sharpens (both columns are derivations from ProxPC ns/day divided by NVIDIA's rated TDP and VRAM, not ProxPC-published). The RTX 5090 hits 0.19 ns/day per watt and 3.44 ns/day per VRAM GB, while the H200 hits 0.19 ns/day per watt and 0.96 ns/day per VRAM GB. Per-watt is a wash. Per-VRAM-GB strongly favors RTX 5090 under 32 GB, and H200 once you need the headroom. SXM-socket silicon bought for AMBER or GROMACS pays for an interconnect the kernel never touches.
From the BIZON Build Floor
Water-cooled BIZON chassis hold sustained GPU clocks through multi-day MD production runs. Equivalent air-cooled four-GPU builds may throttle once the chassis heats up under sustained load, which shows up as reduced aggregate ns/day across a week-long run rather than a hard failure event. The cooling loop, not the GPU SKU, is what separates benchmark ns/day from production ns/day on any four-plus GPU MD rig.
Engine parallelism patterns decide GPU count. SKU tier is a downstream consequence.
Common GPU Mistakes for MD Workloads
The six GPU mistakes below burn through more MD hardware budget than any other pattern in 2026. Four are SKU-and-engine mismatches an engine-first conversation catches at quote stage, and two are reliability traps that surface only after a week on the wrong hardware.

- Buying an H100 for AMBER - RTX 5090 delivers 110 ns/day against the H100's roughly 90. Save the Hopper budget for NAMD at scale.
- Expecting AMBER to scale across four GPUs on one trajectory - It will not. Run four independent trajectories instead.
- Buying 24 GB VRAM for a 50M atom system - It will not fit. You need H200 141 GB or B200 192 GB, and the failure mode is a hard OOM, not a slowdown.
- Buying a Mac for MD - For production GPU-accelerated biomolecular MD, CUDA on NVIDIA remains the safest default. AMBER and NAMD do not run CUDA on Apple Silicon, and although GROMACS 2026 adds an AMD HIP backend for ROCm, Mac is not the right platform for AMBER or NAMD CUDA workflows. See Mac Studio vs NVIDIA GPUs for LLM for where Mac fits.
- Buying SXM silicon for ensemble work - NVLink delivers nothing on independent trajectories. PCIe boards give the same science at a lower silicon tier.
- Skipping ECC for unattended multi-day runs - RTX 5090 is fine for short campaigns. For 72-hour-plus unattended runs, ECC VRAM (RTX PRO 5000 or 6000, H100, H200) protects against bit-flip corruption you do not catch until the analysis stage.
BIZON MD Workstations by GPU Tier
BIZON ships multiple MD workstation tiers in 2026, matched to the engine and atom-count combinations above. Every tier runs BizonOS, our in-house Ubuntu image with NVIDIA drivers, CUDA, and Docker tuned for BIZON hardware. NAMD, GROMACS, LAMMPS, and VMD come preinstalled and tested on top, delivered as NVIDIA GPU-accelerated containers. For CPU, RAM, storage, and cooling, see Best Hardware for Molecular Dynamics Simulations in 2026.
Single-GPU Starting Points
For an individual researcher running AMBER, GROMACS, or OpenMM on systems up to about 1M atoms, a single RTX 5090 is the right starting point. 32 GB of GDDR7 clears the VRAM floor for the overwhelming majority of standard biomolecular simulations at that atom-count range, and ECC VRAM is not required because single-researcher pipelines re-run a corrupted trajectory rather than publishing off it. From our experience at BIZON, this profile fits most PhD students and individual PIs running standard biomolecular MD. Pick by existing lab platform across the AMD and Intel options below.

BIZON X3000 G2 Desktop Workstation
- Best fit - Single-trajectory AMBER, GROMACS, or OpenMM up to about 1M atoms on AMD silicon
- GPUs - Two GPU slots. Compatible with the full RTX Blackwell and Ada workstation lineup (no datacenter SXM or HBM cards).
- VRAM headroom - 32 GB clears the standard biomolecular floor through 1M atoms, and the PRO 6000 option opens 10M+ atom complexes on a single card
- Multi-GPU topology - PCIe Gen 5 x16, no NVLink on workstation Blackwell, so a second card is for parallel trajectories rather than one-trajectory speedup
- ECC - Optional via RTX PRO 6000, not required for single-researcher campaigns that re-run on corruption

BIZON V3000 G4 Desktop Workstation
- Best fit - Same single-trajectory MD profile as the X3000, when the lab is standardized on Intel
- GPUs - Two GPU slots. Compatible with the full RTX Blackwell and Ada workstation lineup (no datacenter SXM or HBM cards).
- VRAM headroom - 32 GB through 1M atoms, with the PRO 6000 option opening 10M+ atom complexes on a single card
- Multi-GPU topology - PCIe Gen 5 x16, second card for parallel trajectories
- ECC - Optional via RTX PRO 6000, not required for single-researcher work
Drug Discovery and Mid-Size Labs
For drug-discovery screening, replica exchange, and GROMACS ensembles, the 2 to 4 GPU tier is where throughput math pays off hardest. ECC becomes worth specifying on multi-day unattended runs, and the RTX PRO 6000's 96 GB of ECC VRAM opens viral-capsid and large-multiprotein work up to about 30M atoms on a single card.

BIZON X5500 G2 Workstation
- Best fit - Flexible mid-tier for labs that mix AMBER, NAMD, and GROMACS across consumer or pro Blackwell silicon
- GPUs - Up to four GPUs. Compatible with the full RTX Blackwell and Ada workstation lineup.
- VRAM headroom - 32 GB tier handles AMBER ensembles to 1M atoms, and the PRO 6000 96 GB tier opens viral-capsid and 10M to 30M atom NAMD on a single card
- Multi-GPU topology - PCIe Gen 5 x16 across all four slots (no NVLink on workstation Blackwell)
- ECC - Available with RTX PRO 6000 for unattended multi-day runs

BIZON G3000 Gen2 Workstation
- Best fit - Reliability-first ECC desktop on Intel for unattended multi-day production runs where bit-flip protection matters
- GPUs - Up to four GPUs. Compatible with the full RTX Blackwell and Ada workstation lineup.
- VRAM headroom - 96 GB per card opens viral-capsid work and 30M+ atom systems on a single GPU
- Multi-GPU topology - PCIe Gen 5 x16 across all four slots, four-way parallel trajectories
- ECC - Full ECC across CPU and GPU, the configuration to choose for week-long unattended runs

BIZON R5000 Rackmount Workstation
- Best fit - The X5500 GPU lineup in a 5U rackmount, for labs with a server room and a queue to replace
- GPUs - Up to four GPUs. Compatible with the full RTX Blackwell and Ada workstation lineup.
- VRAM headroom - Same 32 GB or 96 GB per card as the X5500 tower
- Multi-GPU topology - PCIe Gen 5 x16 across all four slots, drug-discovery screening sweet spot
- ECC - Available with RTX PRO 6000, paired with Threadripper PRO ECC memory

BIZON X8000 G3 Rackmount Server
- Best fit - Hopper PCIe at 4-GPU scale without water cooling, when ECC and 141 GB H200 VRAM matter more than NVLink fabric
- GPUs - Up to four GPUs. MD-relevant options include A100 80 GB, H100 94 GB NVL, and H200 141 GB NVL. RTX PRO and Ada workstation GPUs also supported.
- VRAM headroom - H200 NVL hits 141 GB per card, the PCIe ceiling for whole-cell systems before SXM5 silicon enters the picture
- Multi-GPU topology - PCIe Gen 5 with 2-card NVL bridges (~600 GB/s per pair), not full 8-way fabric
- ECC - Standard on H100 NVL, H200 NVL, and L40S
The hybrid-cooled chassis above handle drug-discovery ensemble screening and periodic production where the four GPUs alternate between active and idle phases across a campaign. For continuous 24/7 max-load production on four consumer cards, step up to the water-cooled ZX5500 in the next section. Hopper PCIe NVL silicon in the X8000 G3 above is engineered for sustained air-cooled operation and does not share that throttle risk.
Department Scale and NAMD HPC (4 to 8 GPU)
For core facilities, NAMD whole-cell work, and mixed-engine production, the 4 to 8 GPU tier is where NVLink fabric earns its premium. Above roughly 30M atoms, only H200 or B200 clears the VRAM floor, and NVLink keeps NAMD scaling near-linear (quantified in the NVLink decision tree above). Cooling and chassis sizing for sustained 8-GPU MD load belong with the system-build conversation, see Best Hardware for Molecular Dynamics Simulations in 2026.

BIZON ZX5500 Water-Cooled Workstation
- Best fit - Water-cooled desktop tower for CPU-heavy GROMACS and LAMMPS pipelines at department scale
- GPUs - Up to seven water-cooled GPUs. Compatible with RTX 5090, RTX PRO 6000, A100 80 GB, H100 80 GB, and H200 141 GB.
- VRAM headroom - Spans the consumer-to-Hopper-PCIe range; H200 option clears the 141 GB whole-cell ceiling
- Multi-GPU topology - PCIe Gen 5 peer-to-peer (no NVLink fabric on workstation Blackwell or H100/H200 PCIe variants)
- ECC and sustained clocks - ECC available with RTX PRO 6000 and H100/H200; full custom liquid keeps clocks pinned 24/7

BIZON ZX9000 Water-Cooled Server
- Best fit - 8-GPU water-cooled server for replica exchange and viral-capsid NAMD where PCIe peer-to-peer scales adequately
- GPUs - Up to eight water-cooled GPUs. MD-relevant options include RTX PRO 6000, A100 80 GB, H100 94 GB NVL, and H200 141 GB NVL.
- VRAM headroom - 141 GB per H200 NVL card clears whole-cell systems past the H100 80 GB cliff
- Multi-GPU topology - PCIe Gen 5 with 2-card NVL bridges, not full 8-way SXM5 fabric
- ECC and sustained clocks - ECC standard on Hopper NVL silicon; enterprise custom liquid sustains full clocks across all 8 GPUs

BIZON X9000 G3 HGX Server
- Best fit - True 8-way SXM5 NVLink fabric for NAMD GPU-Resident Mode above 10M atoms when the NVLink decision tree resolves to yes
- GPUs - Eight H100 80 GB SXM5 or eight HGX H200 141 GB SXM5 (fixed SXM-only chassis).
- VRAM headroom - 80 GB or 141 GB per card; 1.13 TB aggregate on the H200 SXM5 build for whole-cell NAMD
- Multi-GPU topology - Full 8-way NVLink 4 fabric at 900 GB/s per GPU bidirectional, the configuration the NVLink decision tree was written for
- ECC and sustained clocks - ECC standard; HGX 8U air-cooled chassis purpose-built for sustained Hopper SXM5 thermal load

BIZON X9000 G4 HGX Server
- Best fit - Whole-cell NAMD and 30M+ atom systems where Hopper VRAM is no longer enough
- GPUs - Eight NVIDIA B200 192 GB SXM5 (fixed SXM-only chassis, 8 TB/s memory bandwidth per card).
- VRAM headroom - 192 GB HBM3e per card at 8 TB/s memory bandwidth, 1.5 TB aggregate
- Multi-GPU topology - NVLink 5 fabric across all 8 SXM5 modules
- ECC and sustained clocks - ECC standard; HGX 8U air-cooled chassis sized for B200's 1,000W TGP
Getting Started With Your MD GPU
Pick the GPU first against your dominant engine, then size VRAM against your largest production system plus 20 percent headroom. Most labs land on one or four RTX 5090s.
- Engine first - Map your dominant engine using the quick recommendations table.
- Largest production system - Map atom count to a VRAM tier using the VRAM cliff table, plus 20 percent headroom.
- NAMD above 10M atoms? The VRAM tier from step 2 (96 GB+) is required regardless. Use the NVLink decision tree above to choose PCIe peer-to-peer or SXM-NVLink at that tier.
The MD GPU question in 2026 is which card matches your engine's parallelism pattern, and for most labs the answer is a consumer-tier card. Blackwell silicon cuts a production microsecond from weeks to days against Quadro-era hardware, and a four-GPU RTX 5090 workstation runs four such trajectories in parallel for drug-discovery throughput. NAMD whole-cell work above 30M atoms is the one place the math flips toward H200 NVLink silicon. BIZON configures each chassis around the customer's actual workload, with the full driver stack pre-tuned. Browse our MD workstation and server lineup or contact engineering for a custom MD configuration.
BIZON manufactures the workstations and servers discussed in this article. Benchmark figures and product specifications reflect published vendor sources and BIZON build-floor experience. Our editorial recommendations follow the engine-first, workload-first analysis shown above. They are not constrained by inventory or commercial considerations.